OmniScape™ 2025

Understand your target protein with OmniScape™
Optimized for flexibility and ease of use
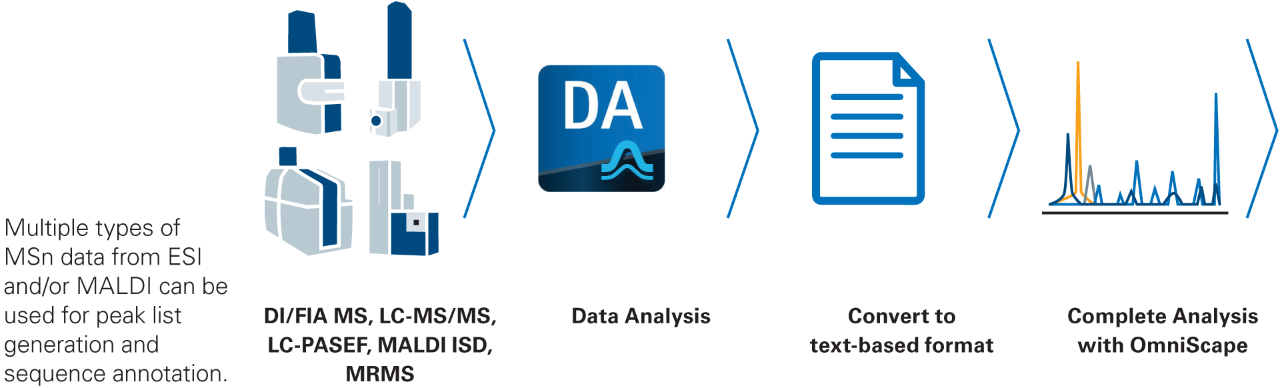


Organizing and analyzing Top-Down MS data can be a challenge. Therefore, bottom-up analysis is often preferred instead of the analysis at the proteoform level. However, characterizing proteins at the intact level offers unique views to understand the biology.
OmniScape™ aims at reducing data handling challenges and bringing Top-Down analysis to the mainstream by tackling typical bottlenecks:
- Top-Down spectra are often complex with a wide range of multiply charged species and a high peak density. OmniScape™ addresses this with the OmniWave™ algorithm to help users with the creation of high-quality peak lists
- Scientists may face a wide range of analytical questions from identifying unknowns to sequence verification of recombinant proteins. OmniScape™ supports de novo analysis combined with homology searches and sequence mapping based on template sequences to assist with these multiple requirements
- Manual validation of individual peaks are cumbersome and can be very time-consuming. With extensive support to quickly verify the quality of the matches OmniScape™ accelerates the analysis for users of high mass accuracy instruments
A single analytical method is often insufficient to capture the complexity of intact proteins including their complex proteoform landscape. OmniScape™ is designed for flexibility and allows users to import data from most ESI or MALDI instrument and/or combine experiments using multiple fragmentation modes.
Designed to meet your Top-Down needs

Users can access two modes of operation depending on the required level of protein characterization, either de novo protein identification or sequence validation with PTMs and other proteoforms.
De novo analysis
Here, the software offers a streamlined procedure (OmniWave™) to calculate sequence tags already formatted for a homology-based search. Fast peak validation and tag generation can save hours of manual effort.
Sequence validation
Once a template sequence is available, either from prior knowledge or a homology search result, OmniScape™ makes it easy to evaluate the data against the sequence including variable modifications. The resulting identifications can be compared to determine the most likely proteoform candidates, making it straightforward to compare results and keep track of the results.
Results combination
Finally, when a single spectrum is not enough to explain the whole sequence, multiple results can easily be merged to enhance the coverage and obtain a high confidence comprehensively annotated identification.
Compatible with MALDI Top-Down sequencing workflows
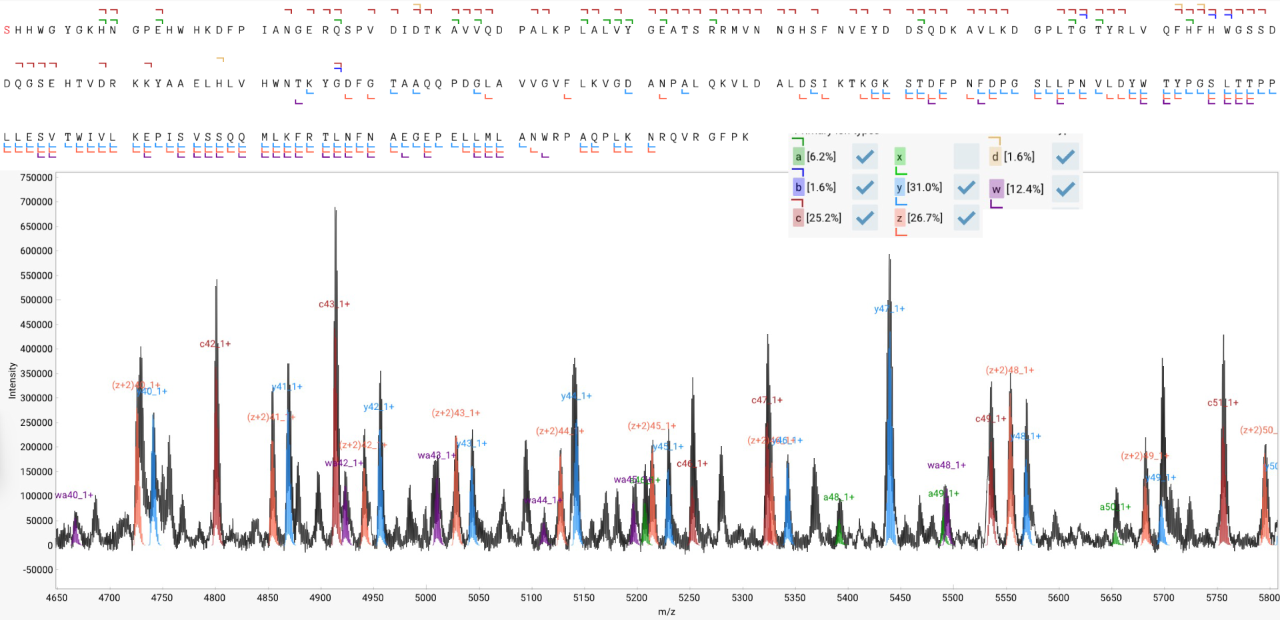
MALDI Top-down sequencing is an extremely fast (data acquisition < 1 second) and efficient way to identify a purified protein including the primary sequence and terminal modifications. This method produces a long series of singly charged fragment ions and affords a nearly instantaneous measurement. Previous studies have shown this approach provides comprehensive sequence coverage for molecules up to 30 kDa.
The OmniWave™ procedure automates the peak finding in MALDI-TDS and automatically extracts monoisotopic masses even for fragments with a low signal to noise.
The software then proceeds to calculate de novo possible sequence tags that explain the measured peaks. These tags are then automatically formatted for homology search to produce a putative identification.
Using the verification workflows the primary sequence and modifications associated with the identified protein can then be tested against the data to improve the sequence coverage and evaluate which proteoform is in the sample.

"The OmniScape™ solution addresses a real need for us in the biotech industry producing recombinant proteins"
Christian Isak Jørgensen, Ph.D., Senior Science Manager, Novonesis, Lyngby, Denmark

"OmniScape™ has intuitive controls that makes fragments annotation and validation in Top-Down MS faster and more accessible. I like how easy it is to hover through the spectra and validate fragment ion matches.
I am excited about the de novo sequencing function, which will help us to discover and annotate proteins in untargeted Top-Down Proteomics runs.
The most impressive function of OmniScape™ is the automatic suggestion of matching proteoforms based on multiple variable modifications."
Boris Krichel, Ph.D., Postdoctoral Fellow, University of Wisconsin-Madison, WI, USA
Literature Room Mass Spectrometry






For Research Use Only. Not for use in clinical diagnostic procedures.