SCiLS™ 软件解决方案
将数据转化为知识
一个软件中完成数据分析和可视化

SCiLS™ 软件
质谱成像的数据处理软件
布鲁克的软件对 MALDI 成像数据的统计分析从未如此简单。
空间可视化并生成分析报告
- 探索每一张谱图的每一个分子特征
- 直接研究分子图像,并与病理学知识相结合
- 快速生成统计学图表及分析报告
- 构建 3D 质谱成像模型 ( 升级选配功能 )
先进的数据处理和分析
- 对样本数据无限制的先进机器学习算法
- 差异性比较分析用于发现潜在的生物标志物
- 无监督的空间分割及主成分分析功能用于非靶向的聚类及数据挖掘
- 对区域进行多重信息注释以对接临床科研
- 用于非标记样本的分类和模型建立
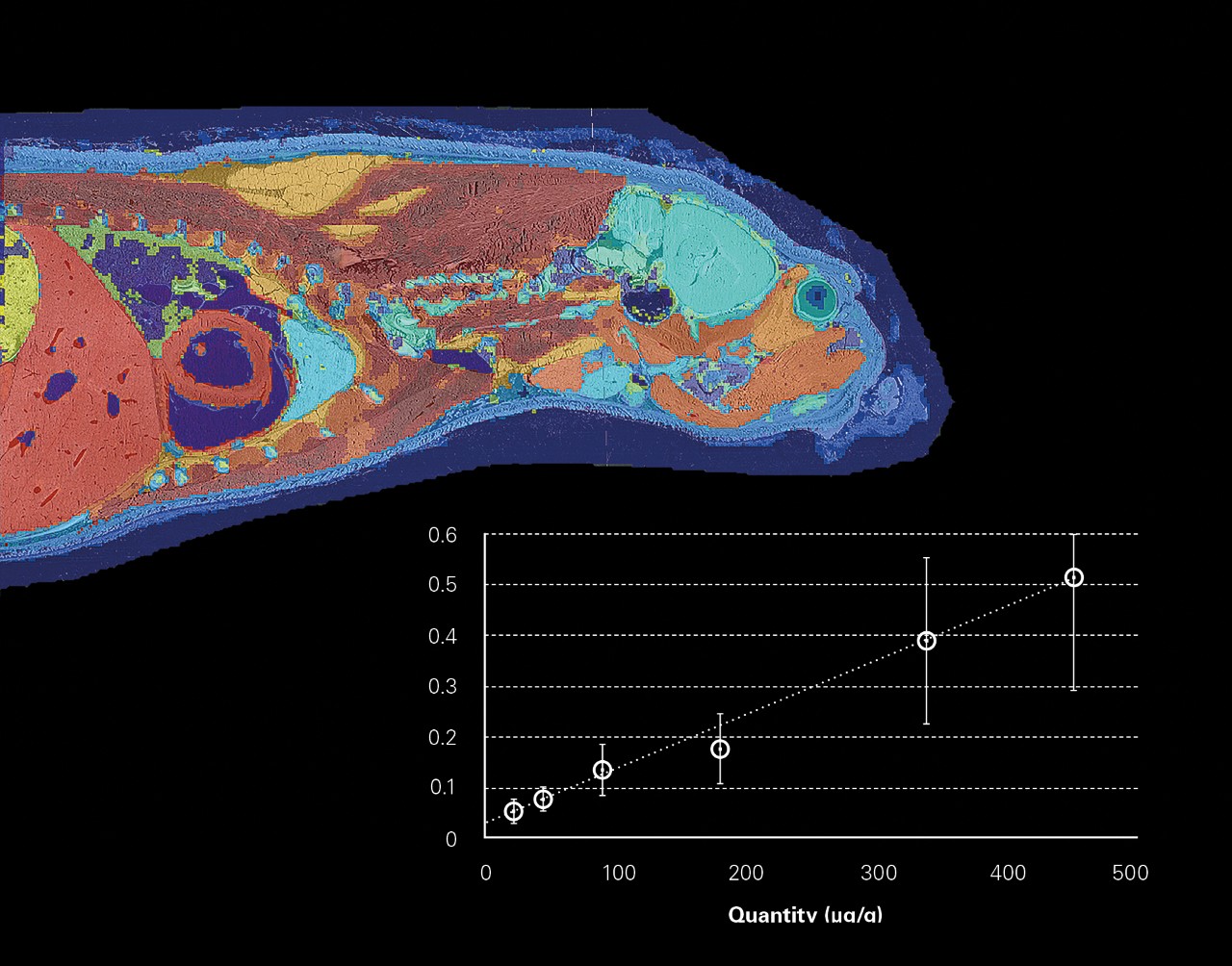
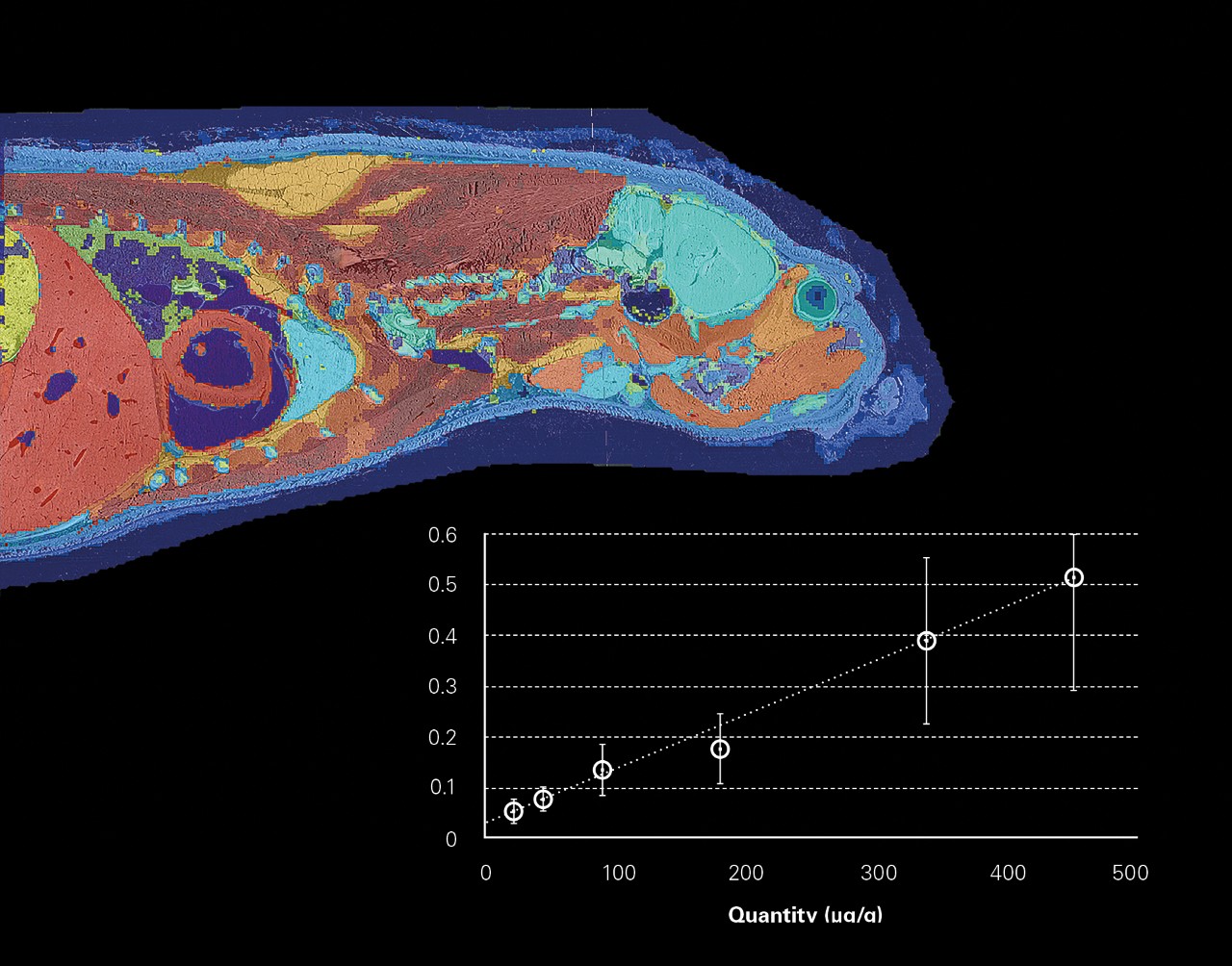
- 梯度稀释法用于目标分子的质谱成像绝对定量分析
工作流程及可拓展性
- 集成了多种统计分析工具,适配于不同的靶向及非靶向空间定位组学工作流程 ( SpatialOMx® )
- 与病理组织学的标注结果无缝衔接
- 编程语言接口用于进一步功能拓展,如自动生成报告及多平台数据整合等
- 质谱成像的数据可以导出成通用的 imzML 格式
- 可以导入 imzML 格式、或其他品牌仪器的质谱成像数据进行后续的统计分析(升级选配功能)

系统要求
数据系统硬件要求:
- 英特尔酷睿 i7 处理器或英特尔 Xeon 处理器 ( 相同或更高 ) ,至少为四核处理器
- 微软 Windows 10 64 位操作系统
- 64 GB 内存 ( 对于大量样本或大体积数据,推荐使用 128 GB 内存 )
- 1 TB 的硬盘空间,推荐使用 SSD 固态硬盘,更多信息见下方
- 全高清显示屏 ( 1920 x 1080 )
- 支持 OpenGL 3.2 的显卡,GPU 容量至少 4 GB
为了实现最优性能,推荐使用 SSD 固态硬盘来存放 SCiLS™ Lab 文件。
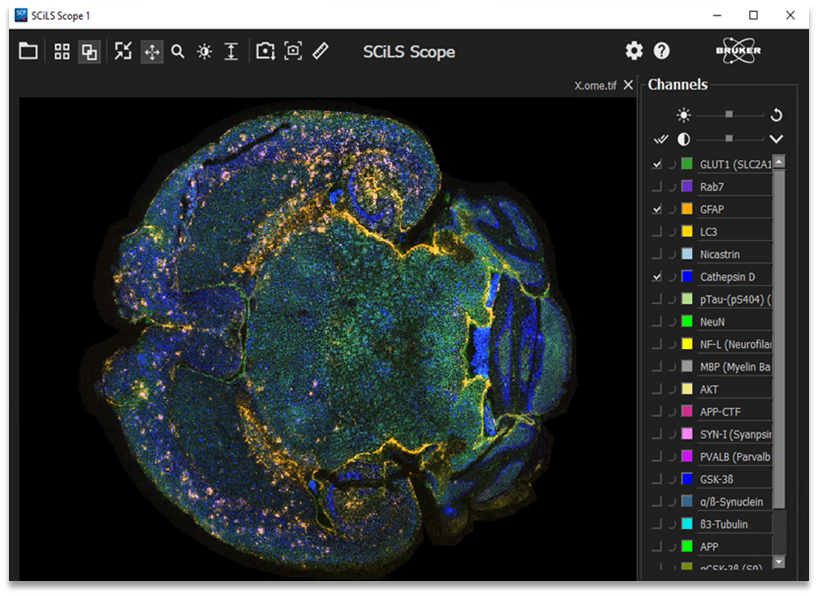
全新的可视化软件:专门用于 MALDI HiPLEX-IHC 成像数据的共享及空间可视化
SCiLS™ Scope 专为 MALDI 成像数据和 MALDI HiPLEX-IHC 成像数据的共享及空间可视化而设计。界面操作简单、易于使用,采用开放式的 OME-TIFF 格式的图像文件,便于与合作者进行科研共享。可以直观地与病理学标注结果进行快速比对,以实现单个像素点信息挖掘的最大化。
MALDI HiPLEX-IHC 成像数据的空间可视化
SCiLS™ Scope 的比对功能包括上下重叠,精细调整以及单个分子通道的独立分析。它给多个靶向蛋白质的独立及整合分析提供了很好的切入点。
SCiLS™ Scope 提供了多种分析功能,用于快速检视 MALDI HiPLEX-IHC 靶向蛋白质组学数据。每个抗体对应的质量标签都可以单独分析、或整合分析。此外,该软件还具有其他的特色功能:
- 连续的放大功能
- 标尺功能:对生物学相关结构的测距
- 图像导出:复制到剪切板或另存为 TIFF 等格式
- 颜色及对比度调整:对单个分子通道做针对性调整,或对所有分子通道做宏观调整
与 MALDI HiPLEX-IHC 工作流程的深度整合
SCiLS™ Scope 是靶向蛋白质组学 MALDI HiPLEX-IHC 工作流程的重要环节。SCiLS™ autopilot 自动一体化数据采集流程使用默认的采集方法,并利用已知的 HiPLEX 质量标签进行实时校准以获取高质量的质谱数据。SCiLS™ Lab 可以实现后续的数据自动导入、预处理及 OME-TIFF 图像结果的自动导出,为分析流程提供立等可取的结果。
系统要求
以下是推荐的系统硬件要求:
- 硬盘:至少 100 GB 的可用磁盘空间。
- 主存储器:至少 32 GB RAM。
- 操作系统:微软 Windows 10 64 位。
- 图形处理器:AMD Radeon 或 NVIDIA 高端图形处理器,4 GB 显存。
- 图形分辨率:1920 x 1024 分辨率,真彩色。
世界领先的质谱成像数据软件
SCiLS™ Lab 可处理所有主要质谱仪的数据,包括布鲁克的 FLEX 系列和 MRMS 系列,以及 imzML 开放格式数据。对于 timsTOF fleX 产生的淌度成像数据,SCiLS™ Lab 展示质量-离子淌度热的二维热图,并实现 CCS 数据的深度解析。
多个样品的比较分析可以通过二维和三维空间中的可视化来实现,应用于 多个领域,例如药物研发、组织的特征性标志物的发现、补充免疫组化的临床病理学等方面的研究。
SCiLS™ Lab 支持对组织中的目标分子进行定量质谱成像分析。SCiLS™ Lab 与 MetaboScape® 对接、整合于空间定位组学工作流程 ( SpatialOMx® ),将 MALDI 成像检测到的代谢物、脂质、多糖等特征分子可视化并产生分子信息注释。SCiLS™ Lab与开源的 QuPath 软件无缝对接,可实现病理标注与分子成像的整合分析。

无标记分子成像质谱覆盖蛋白质到小分子
SCiLS™ Lab 可用于分析一个或多个 MALDI 成像数据集,以平铺方式显示多个数据集并在空间上对齐。可视化和详细的统计分析不受数据集多少的限制。此外,SCiLS™ Lab 还可用于连续 2D 数据集 MALDI 成像后的 3D 图像重建,以及 3D 成像数据的可视化和分析。


大队列临床病理学样本
在生物标志物发现中 ,寻找区分离子往往通过手动或自己编写软件完成。 由于生物标志物研究中要分析的样本数量和数据量巨大,手动数据分析既耗时又容易出错。使用SCiLS™ Lab 可以轻松解析谱图,区分病理生理变化。
现在只需单击一下,SCiLS™ Lab 即可自动评判所有离子对不同生物状态的区分能力。可能的特征生物标志物被列表输出用于后续分析。
使用 SCiLS™ Lab,只需单击一下可以发现强大的数据挖掘功能。
整体成像
2006 年开创性的全身 MALDI 成像,对方法学和数据计算是一个挑战。为了可视化药物和代谢物在整体的组织分布,使用各种商业、学术和实验室自开发软件完成了数据的分布处理。
现在一个软件可以对由数百万个谱图组成的数据集进行所有处理。从多个数据集注册到一个分析文件,到谱图的预处理,再到综合统计分析,SCiLS™ Lab 提供了数据分析的完整工作流程。
相关网络讲座

“ 借助 SCiLS Lab,我们的团队节省了无数小时的手动比较时间。我们热切期待这一独特的功能。”
Rita Casadonte 博士,Proteopath 有限责任公司




质谱文献室



*仅供研究使用,不能用于临床诊断程序。